


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
 anion
anion高柳 敏幸; 和田 晃
Chemical Physics Letters, 348(1-2), p.514 - 520, 2001/11
被引用回数:14 パーセンタイル:41.25(Chemistry, Physical)時間に依存しない量子反応性散乱理論を用いてF(HD)アニオンの光電子脱離スペクトルの計算を行った。StarkとWernerの作製した高精度のポテンシャル面を使った。計算したFHD及びFDH両アニオンのスペクトルには、束縛回転に相当するブロードなピークがいくつか見られた。これは、以前研究されたFH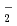 のスペクトルで見られたものと本質的に同じである。さらに、FHDアニオンでは、遷移状態共鳴に相当するピークが見られた。これは、最近、詳細な反応断面積の測定によって実験的に見出されているものである。本理論計算結果は光電子脱離スペクトル実験によって、遷移状態共鳴が見出される可能性があることを強く示唆するものである。
のスペクトルで見られたものと本質的に同じである。さらに、FHDアニオンでは、遷移状態共鳴に相当するピークが見られた。これは、最近、詳細な反応断面積の測定によって実験的に見出されているものである。本理論計算結果は光電子脱離スペクトル実験によって、遷移状態共鳴が見出される可能性があることを強く示唆するものである。
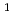 D)+N
D)+N O
O NO+NO reaction
NO+NO reaction高柳 敏幸; 和田 晃
Chemical Physics, 269(1-3), p.37 - 47, 2001/07
被引用回数:14 パーセンタイル:41.25(Chemistry, Physical)O(D)+NONO+NO反応について、量子反応性散乱計算を行った。ポテンシャルエネルギー曲面は、CASPT2レベルの高精度の分子軌道計算を行い、解析関数にフィットして作製した。反応側及び生成側の配向角を固定したモデルを用いることによって、次元を3次元に落とした。この反応では2種類のNO分子が生成する。反応熱はおもに新しく生成するNO分子の振動に分配されるが、もともと存在したNO振動モードにも、ある程度エネルギーが分配されることを見出した。このことはもともと存在したNO結合が、必ずしもスペクテータではないことを示している。
 P
P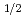 )+H HBr+H reaction
)+H HBr+H reaction高柳 敏幸; 黒崎 譲
Journal of Chemical Physics, 113(17), p.7158 - 7164, 2000/11
被引用回数:43 パーセンタイル:77.61(Chemistry, Physical)スピン軌道相互作用による電子的非断熱遷移を伴う反応、Br(P)+H HBr+Hについて3次元量子反応性散乱計算を2つの計算方法を用いて行った。1つは超球座標を用いたclose-coupling法で、もう一方は、虚数の吸収ポテンシャルを用いた一般化R行列伝播法である。後者では反応側のJacobi座標を用いた。ポテンシャル曲面としてはTruhlarらによる(2 2)のdiabaticなポテンシャル曲面を用いた。いずれの方法でも数値的に十分収束した計算結果を得ることができた。また、得られた結果から電子的非断熱遷移が反応の入口でほとんど起こるが、その効率は小さいことがわかった。
2)のdiabaticなポテンシャル曲面を用いた。いずれの方法でも数値的に十分収束した計算結果を得ることができた。また、得られた結果から電子的非断熱遷移が反応の入口でほとんど起こるが、その効率は小さいことがわかった。
HCN+H reaction; Applications of the rotating-bond approximation高柳 敏幸; G.C.Schatz*
Journal of Chemical Physics, 106(8), p.3227 - 3236, 1997/02
被引用回数:50 パーセンタイル:84.11(Chemistry, Physical)Rotating-Bond近似(RBA)をH+HCN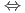 CN+H反応に適用した。CNの振動モードを計算に取り入れた。解くべき4次元のシュレディンガー方程式は起球座標と離散変数表示法を組み合わせて数値的に解いた。CH振動モードとCNの振動モードの結合が反応ダイナミクスに大きな影響を与えることがわかった。反応のしきい値は初期振動エネルギーのみで決まることがわかった。このことは反応に直接関与しないと考えられるCN振動やHCNの変角振動も反応座標と結合していることを示す。しかし、H+HCN反応についての断面積はC-H振動の励起によってかなり増大する。これはこの反応がモード特異的であることを示すものであり、実験結果とも定性的に一致する。
CN+H反応に適用した。CNの振動モードを計算に取り入れた。解くべき4次元のシュレディンガー方程式は起球座標と離散変数表示法を組み合わせて数値的に解いた。CH振動モードとCNの振動モードの結合が反応ダイナミクスに大きな影響を与えることがわかった。反応のしきい値は初期振動エネルギーのみで決まることがわかった。このことは反応に直接関与しないと考えられるCN振動やHCNの変角振動も反応座標と結合していることを示す。しかし、H+HCN反応についての断面積はC-H振動の励起によってかなり増大する。これはこの反応がモード特異的であることを示すものであり、実験結果とも定性的に一致する。
 H+CH
H+CH reaction
reaction高柳 敏幸
Journal of Chemical Physics, 104(6), p.2237 - 2242, 1996/02
被引用回数:97 パーセンタイル:94.08(Chemistry, Physical)H+CHH+CH反応について次元を落とした量子反応性散乱理論を用いて調べた。系を直線4原子の反応として取り扱い、数学的な次元を3次元にまで少なくした。振動モードとしては、CHの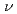
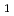 およびモード、Hの振動、CHのかさ振動であるモードが考慮された。ポテンシャルエネルギー曲面としてはJordanとGilbertによる半経験的ポテンシャル関数を用いた。回転平均した反応断面積および反応速度定数はエネルギーシフト近似を用いて計算した。計算の結果、CHのモードの励起が反応性に著しく影響を与えることがわかった。これは反応座標とモードのカップリングが強いことを示している。またHとCHの振動分布について調べたところ、Hはあまり振動励起していないが、CHのモードは反応によって励起していることがわかった。
およびモード、Hの振動、CHのかさ振動であるモードが考慮された。ポテンシャルエネルギー曲面としてはJordanとGilbertによる半経験的ポテンシャル関数を用いた。回転平均した反応断面積および反応速度定数はエネルギーシフト近似を用いて計算した。計算の結果、CHのモードの励起が反応性に著しく影響を与えることがわかった。これは反応座標とモードのカップリングが強いことを示している。またHとCHの振動分布について調べたところ、Hはあまり振動励起していないが、CHのモードは反応によって励起していることがわかった。
D)+H reaction高柳 敏幸; 小林 浩信*; 綱島 滋*
J. Chem. Soc., Faraday Trans., 92(8), p.1311 - 1314, 1996/00
N(D)+H反応のダイナミクスについて正確な量子散乱理論および古典的トラジェクトリー法を用いて調べた。ポテンシャルエネルギー曲面としては、最近ab initio分子軌道計算をもとに作製された関数を用いた。量子計算については超球座標系を用いた。また計算は全角運動量J=0でのみ行った。反応断面積および反応速度定数を求めるのにJ-シフト近似を用いた。量子計算および古典計算ともに生成物のNHの振動分布は実験値をよく再現することがわかった。反応速度定数については量子計算と実験値はよく一致する。しかし一方、古典的トラジェクトリ法はかなり大きな反応速度定数を与えることがわかった。
